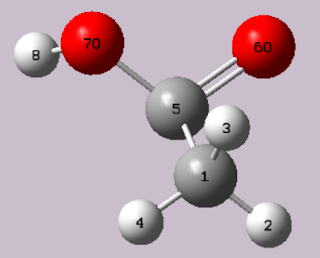
如何用Gaussian確認物質結構及作IR分析

在上一篇文章, 如何用Gaussian看波函數 ,我提到 注意!此分子的波函數不會是現實中分子的波函數,因為他可能結構有異,或是因為在現實中他是在水溶液環境中,(Gaussian預設為在真空中氣體分子的互動)。 其實在文章中,你也能很明顯看出COOH鍵結的不尋常角度。 那我們要怎麼優化化學分子的結構呢? 1. 用GaussView開啟想要看的分子,chk檔,GJF檔,LOG檔皆可 2. 然後 點擊Gaussian calculation setup(快捷ctrl+G) 3. 選取Opt/Freq,即Optimization&F requency,然後點選左下角的Submit,後面參數先不用調整。 4. 等待G09計算,計算完成即可關閉G09,此時開啟chk檔 左側log檔為,優化後的結構,就是分子在氣態下實際的長相。 中間gjf檔為,原先自行繪出的結構 右側chk檔為,優化過後的結構且保留運算方法,這個才是IR分析要的! 觀察磁碟大小,也可以明顯發現chk檔檔案較大 5. 關閉gjf檔與log檔,對著chk檔,選取Results>Vibrations 此視窗,直接點選OK即可 IR圖與頻譜分析就出來囉! 圖中的振動頻率分析相當重要,因為G09本身計算問題,所以可能導致,即使是優化過了,仍會輸出非穩定且具有虛頻的結構,唯有頻律皆存在的才是物質穩定的結構。 那該如何調整結構至穩定態呢?請待後續文章揭曉 此處,可以點Start Animation觀看分子震動動畫。可以加速、加振福或是顯示向量。 不說啦!我先抖一波啦! 注意!此處的數據仍會與現實產生誤差,因為溶液環境還沒調整過。 那該如何調整環境呢?請待後續文章揭曉。 若是您正在研究量子化學或是有興趣的話,歡迎繼續點閱後續的文章。 敬請不吝指教!! 參考: 1.Gaussian官網: http://gaussian.com/ 2.成大化學系 鄭沐政老師 - 分子與材料模擬技術課程