如何用Gaussian看波函數
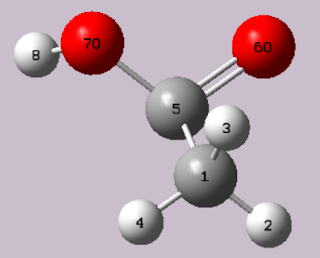
Gaussian可以拿來觀看分子的波函數,可以選擇要看的是LUMO,HOMO,或是總電子雲密度。 1. 創建分子團 首先,用GaussView創建新的分子團,角度跟方向先不用刻意處理,只要分子相對位置及鍵結差不多即可! 此處以乙酸為例 2. ctrl+s存檔,並以記事本打開此檔案,即可檢視 。 圖中會顯示chk檔位置,使用方法,電荷,多重度,座標,鍵結級數 3. 回到GaussView視窗,點擊Gaussian calculation setup(快捷ctrl+G) 4. 選取能量,然後點選submit,後面參數先不用調整 5. 等待G09計算,直到執行完成,即可先關閉G09, 此視窗請點選.chk,不然看不到波函數 計算完成的分子,乍看下是一樣的,但是上面附檔名會是.chk 6. 回到GaussView視窗,點選Result>Surfaces/Contours 在此畫面中,選取New Cube 這裡可以選取顯示的有,MO、電子雲密度 在MO中,可以選取LUMO、HOMO,或是一次選取 生成後,選取Cube,再點選New Surface,就可以看到選定的MO了 乙酸HOMO 乙酸LUMO 要看總電子密度也是可以的 選取Mapped Surface可以看到密度分布 選取只顯示表層,然後選ESP 表層電子雲密度分布就出現了 選取Hide,可以隱藏掉不要顯示的部分喔! 回到GaussView,選取Save image file或是Windows+S,就能保存當前2D畫面囉! 那要怎麼保存3D圖呢?那請把chk檔給想要看的人吧! 注意!此分子的波函數不會是現實中分子的波函數,因為他可能結構有異,或是因為在現實中他是在水溶液環境中,(Gaussian預設為在真空中氣體分子的互動)。 那該如何調整或優化結構,或是調整環境呢?請待後續文章揭曉。 若是您正在研究量子化學或是有興趣的話,歡迎繼續點閱後續的文章。 敬請不吝指教!! 參考: 1.Gaussian官網: http://gaussian.com/ 2.成大化學系 鄭沐政老師 - 分子與材料模擬技術課程